
A Python package to automate the analysis of potential hydrogen bonds and similar type of weak interactions like halogen bonds and non-canonical interactions in macromolecular structures, available in Brookhaven Protein Database (PDB) file format. HBAT uses a geometric approach to identify potential hydrogen bonds by analyzing distance and angular criteria between donor-hydrogen-acceptor triplets.












HBAT v2 is a modern Python re-implementation of the original Perl-based tool developed by Abhishek Tiwari and Sunil Kumar Panigrahi.
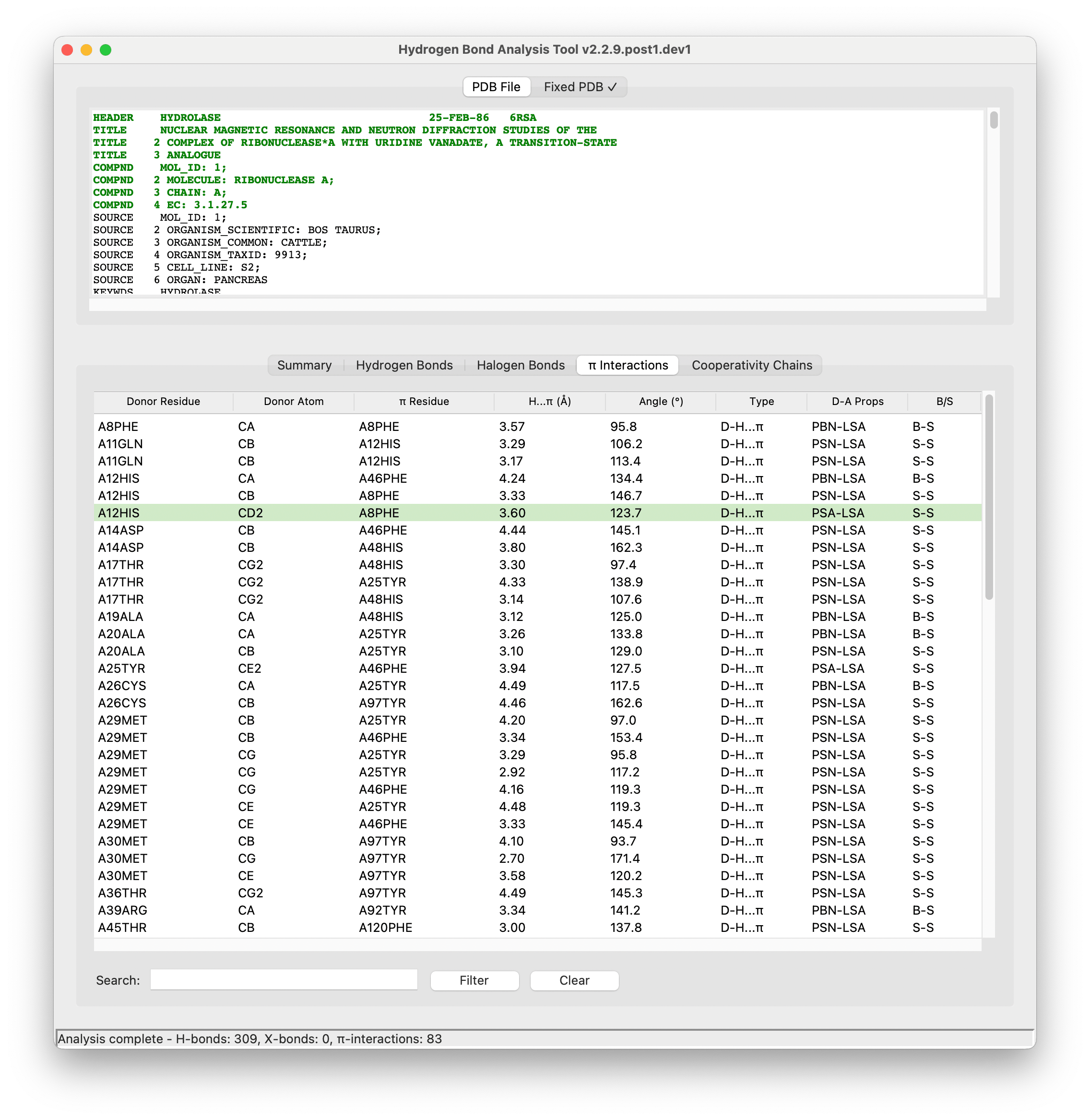
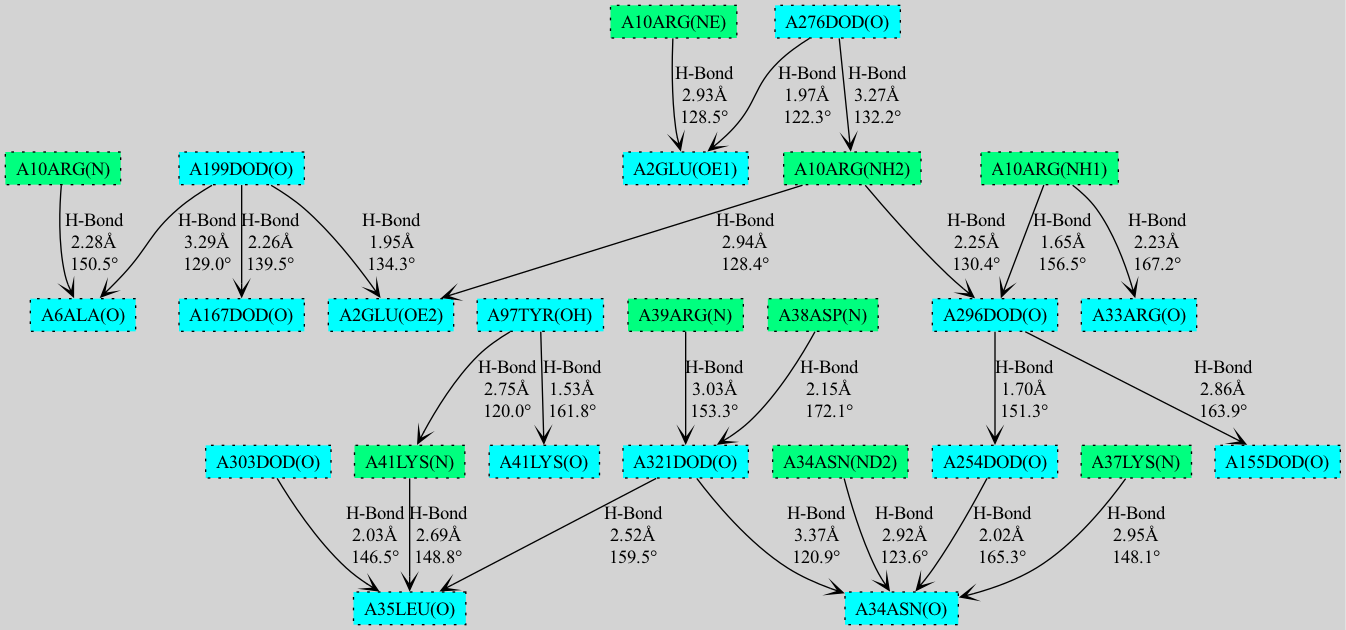
- Comprehensive Analysis: Detect and analyze potential hydrogen bonds, halogen bonds, and X-H...π interactions
- Dual Interface: Both graphical (tkinter) and command-line interfaces
- Advanced Visualization: Choice between NetworkX/matplotlib and GraphViz renderers for cooperativity chain visualization
- High-Quality Graphics: Export visualizations to PNG, SVG, PDF with configurable resolution
- Interactive GUI: Scrollable visualizations with dynamic layout switching and engine selection
- Parameter Presets: Built-in presets for different structure types (high-resolution, NMR, membrane proteins, etc.)
- Flexible Parameters: Customizable distance cutoffs, angle thresholds, and analysis modes.
- Multiple Output Formats: Text, CSV, and JSON export options
- Fast Processing: Optimized algorithms for efficient analysis of large structures
- Cross-Platform: Works on Windows, macOS, and Linux.
Please review HBAT documentation for more details.
{kind=link}
{kind=link}
- Hydrogen Bonds: O-H...O, N-H...O, N-H...N, and other X-H...Y interactions
- Halogen Bonds: C-X...Y interactions (X = F, Cl, Br, I; Y = N, O, S)
- X-H...π Interactions: Hydrogen bonds to aromatic ring systems
Please review HBAT documentation for more details.
pip install hbatRun HBAT Command-Line Interface (CLI) using hbat or launch HBAT GUI using hbat-gui.
git clone https://github.com/abhishektiwari/hbat.git
cd hbat
pip install -e .Alternatively,
pip install git+https://github.com/abhishektiwari/hbat.gitRun HBAT Command-Line Interface (CLI) using hbat or launch HBAT GUI using hbat-gui.
conda install -c hbat hbat
- Python: 3.9 or higher
- tkinter: tkinter is included with Python standard library on most systems. However, on Mac install Python and tkinter using
brew.
brew install python python3-tk
- GraphViz (Optional): Required for advanced cooperativity chain visualization with high-quality graph rendering. HBAT will automatically fall back to NetworkX/matplotlib visualization if GraphViz is not available.
Install GraphViz:
On Ubuntu/Debian:
sudo apt-get update
sudo apt-get install graphvizOn macOS (using Homebrew):
brew install graphvizOn Windows:
- Download and install from GraphViz official website
- Or using Chocolatey:
choco install graphviz - Or using conda:
conda install -c conda-forge graphviz
Note: After installing GraphViz, restart your terminal/command prompt before running HBAT to ensure the GraphViz executables are available in your PATH.
Launch the GUI application:
hbat-guiThe GUI provides,
- File browser for loading PDB files
- Parameter configuration panels
- Tabbed results display
- Export and visualization options
Basic usage:
hbat input.pdbHBAT supports multiple output formats with automatic detection based on file extension:
# Single file outputs (format auto-detected from extension)
hbat input.pdb -o results.txt # Text format
hbat input.pdb -o results.csv # CSV format (single file with all data)
hbat input.pdb -o results.json # JSON format (single file with all data)
# Multiple file outputs (separate files per interaction type)
hbat input.pdb --csv results # Creates results_h_bonds.csv, results_x_bonds.csv, etc.
hbat input.pdb --json results # Creates results_h_bonds.json, results_x_bonds.json, etc.With custom parameters:
hbat input.pdb -o results.csv --hb-distance 3.0 --mode localhbat --list-presetshbat protein.pdb --preset high_resolution
hbat membrane_protein.pdb --preset membrane_proteinshbat protein.pdb --preset drug_design_strict --hb-distance 3.0 --verbosepositional arguments:
input Input PDB file
optional arguments:
-h, --help show this help message and exit
-o OUTPUT, --output OUTPUT
Output file (format auto-detected from extension: .txt, .csv, .json)
--json JSON Export to multiple JSON files (base name for files)
--csv CSV Export to multiple CSV files (base name for files)
Preset Options:
--preset PRESET Load parameters from preset file (.hbat or .json)
--list-presets List available example presets and exit
Analysis Parameters:
--hb-distance HB_DISTANCE
Hydrogen bond H...A distance cutoff in Å (default: 3.5)
--hb-angle HB_ANGLE Hydrogen bond D-H...A angle cutoff in degrees (default: 120)
--da-distance DA_DISTANCE
Donor-acceptor distance cutoff in Å (default: 4.0)
--xb-distance XB_DISTANCE
Halogen bond X...A distance cutoff in Å (default: 4.0)
--xb-angle XB_ANGLE Halogen bond C-X...A angle cutoff in degrees (default: 120)
--pi-distance PI_DISTANCE
π interaction H...π distance cutoff in Å (default: 4.5)
--pi-angle PI_ANGLE π interaction D-H...π angle cutoff in degrees (default: 90)
--covalent-factor COVALENT_FACTOR
Covalent bond detection factor (default: 1.2)
--mode {complete,local}
Analysis mode: complete (all interactions) or local (intra-residue only)
Output Control:
--verbose, -v Verbose output with detailed progress
--quiet, -q Quiet mode with minimal output
--summary-only Output summary statistics only
Analysis Filters:
--no-hydrogen-bonds Skip hydrogen bond analysis
--no-halogen-bonds Skip halogen bond analysis
--no-pi-interactions Skip π interaction analysis
This project is licensed under the MIT License - see the LICENSE file for details.
If you use HBAT in your research, please cite:
@software{tiwari2025hbat,
author = {Tiwari, Abhishek},
title = {HBAT: Hydrogen Bond Analysis Tool},
version = {v2},
year = {2025},
url = {https://github.com/abhishektiwari/hbat}
}
@article{tiwari2007hbat,
author = {Tiwari, Abhishek and Panigrahi, Sunil Kumar},
doi = {10.3233/ISI-2007-00337},
journal = {In Silico Biology},
month = dec,
number = {6},
title = {{HBAT: A Complete Package for Analysing Strong and Weak Hydrogen Bonds in Macromolecular Crystal Structures}},
volume = {7},
year = {2007}
}
See our contributing guide and development guide. At a high-level,
- Fork the repository
- Create a feature branch
- Make your changes
- Add tests if applicable
- Submit a pull request