pdb2reaction: End-to-End Reaction-Path Elucidation from PDB Structures Using Machine-Learning Interatomic Potentials
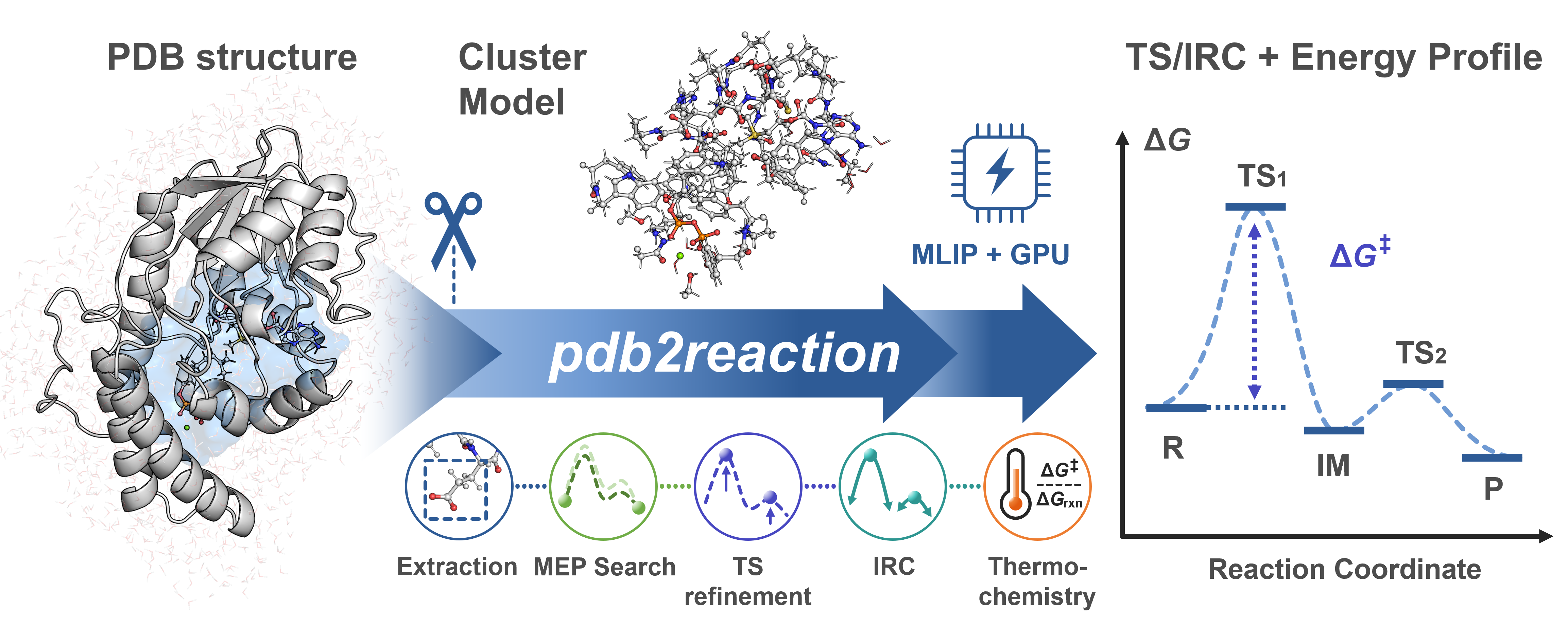
pdb2reaction is a Python CLI for elucidating enzymatic reaction pathways from PDB or mmCIF structures using machine-learning interatomic potentials (MLIPs). Given (i) two or more reaction-ordered structures, (ii) one structure with --scan-lists, or (iii) one TS candidate with --tsopt, it can run an MEP search and optionally chain TS optimization → IRC → thermochemical correction → DFT single-point. Active-site extraction is performed only when -c/--center is supplied; otherwise the PDB/mmCIF/XYZ/GJF model is used as-is. Each stage is also exposed as an individual subcommand.
Test a reaction mechanism in a single command:
# Multi-PDB mode (R + P endpoints → MEP, with TS optimization + thermo)
pdb2reaction all -i R.pdb P.pdb -c 'LIG' -l 'LIG:-1' --tsopt --thermoInputs are not limited to full enzyme PDBs: mmCIF is accepted directly, including multi-character chains and large residue IDs. You can also pass a small molecule as .xyz / .gjf, or a cluster model you built yourself as PDB/mmCIF, and omit --center/-c to skip extraction.
Prerequisites: PDB/mmCIF inputs must already contain hydrogens; reaction-ordered structures must share the same atom identities and order (only coordinates differ). Small-molecule
.xyz/.gjfinputs work when--center/-cand--ligand-charge/-lare omitted.
| Tool | Use case |
|---|---|
| mlmm-toolkit | ML/MM ONIOM with the full protein environment; automates MM parameterization and ML-region assignment from a single PDB. |
| uma_pysis | Lightweight YAML-driven UMA–pysisyphus interface for quick/exploratory reaction-mechanism studies (GS / TS / IRC / ΔG). |
pdb2reactionbundles a GPU-optimized pysisyphus fork that is not compatible with upstream pysisyphus — do not install it into an environment that already has upstream pysisyphus.
- Getting Started · mmCIF and large structures · Installation · Examples · Troubleshooting
- YAML Reference · JSON Output Schema
- Full site: https://t-0hmura.github.io/pdb2reaction/
An interactive Colab notebook is available.
| Component | Requirement |
|---|---|
| OS / Python | Linux recommended. Python >= 3.11. |
| GPU / CUDA / VRAM | CUDA-capable NVIDIA GPU recommended for production, with a compatible driver and official PyTorch 2.8 CUDA wheel (cu126, cu128, or cu129). Required VRAM is backend/model/system/workflow dependent; pilot the real calculation. |
| RAM / Disk | Size for the selected environment, model cache, structures, trajectories, and DFT scratch; no atom-count-only minimum is reliable. |
CPU-only execution works but is usually much slower; benchmark the selected backend/model. Full requirement and tuning details: docs/installation.md.
# 1. CUDA-enabled PyTorch (choose the official 2.8 wheel for your driver/GPU)
pip install 'torch==2.8.0' --index-url https://download.pytorch.org/whl/cu126
# 2. pdb2reaction (editable from a local clone, or `pip install pdb2reaction`)
pip install -e .
# 3. Authenticate Hugging Face once (only required for the default UMA backend)
# Accept the FAIR Chemistry License v1 at https://huggingface.co/facebook/UMA, then:
hf auth login # interactive
# OR: export HF_TOKEN=hf_xxx && hf auth login --token "$HF_TOKEN" --add-to-git-credential # CI / HPCOptional extras (install only what you need):
| Extra | Adds |
|---|---|
[orb] / [aimnet] |
Orb / AIMNet2 MLIP backend (-b orb / -b aimnet2) — not HF-gated |
[dft] |
PySCF + GPU4PySCF single-point DFT (--dft / pdb2reaction dft) |
[mcp] |
Model Context Protocol server for agent clients |
The MACE backend (-b mace) is not a pip extra: mace-torch pins e3nn==0.4.4, which conflicts with fairchem-core's e3nn>=0.5 (UMA), so it needs a dedicated environment — pip uninstall -y fairchem-core && pip install mace-torch (see docs/installation.md).
CUDA module loads, alternative-backend recipes, DMF/cyipopt setup, Plotly Chromium, and HPC job-script templates: docs/installation.md and docs/hpc-example.md.
Examples use GPP C6-methyltransferase BezA (Tsutsumi et al., Angew. Chem. Int. Ed. 2022, 61, e202111217) — runnable MEP and scan commands are in examples/run.sh.
# Multi-structure MEP (R + P → MEP, with TS + thermochemistry)
pdb2reaction -i 1.R.pdb 3.P.pdb -c 'SAM,GPP,MG' -l 'SAM:1,GPP:-3' \
--tsopt --thermo --out-dir result_mep
# Scan mode (single structure → staged bond scan → MEP)
pdb2reaction -i 1.R.pdb -c 'SAM,GPP,MG' -l 'SAM:1,GPP:-3' \
-s '[("CS1 SAM 320","GPP 321 C7",1.60)]' --tsopt --thermo --out-dir result_scan
# TS-only validation (single TS candidate → tsopt → IRC → freq)
pdb2reaction -i TS_candidate.pdb -c 'SAM,GPP,MG' -l 'SAM:1,GPP:-3' --tsopt --thermo --out-dir result_tsonlypdb2reaction can also be used to investigate reaction mechanisms of small molecules and user-defined cluster models.
# Small molecule (gas-phase): .xyz / .gjf input — omit -c, set charge with -q
pdb2reaction -i reactant.xyz product.xyz -q 0 --tsopt --thermo --out-dir result_small
# Your own cluster model (already-trimmed PDB): omit -c to use it as-is
pdb2reaction -i cluster_R.pdb cluster_P.pdb -q 0 --tsopt --thermo --out-dir result_clusterFor hand-built clusters, standardize backbone ends at Cα (CA), place other
boundaries on aliphatic C–C single bonds whenever possible, avoid cutting
peptide/polar/conjugated/metal bonds, and use the identical atom order and cap
topology for every state. See the cluster-boundary checklist.
Per-stage walkthrough (extract → opt → path-opt → tsopt → irc → freq → dft): docs/getting-started.md and docs/quickstart-all.md.
A non-dry all run writes the deliverables reached by its enabled stages to
--out-dir (default ./result_all/):
segments/seg_NN/{reactant,ts,product}.*— the canonical R / TS / P structures to citemep_trj.xyz(plusmep.pdbwhen topology is available andmep.ciffor bridged inputs) — the merged reaction path in MEP/scan-list modesenergy_diagram_MEP.png— MEP diagram when MEP construction and static-image export succeedsummary.log(human-readable) /summary.json(machine-readable)
Pipeline scratch lives under _work/ (safe to delete). Full layout and filename conventions: docs/output-layout.md.
| Subcommand | Role | Doc |
|---|---|---|
all (default) |
End-to-end: extract → MEP → TS → IRC → freq → DFT | all |
extract |
Build active-site cluster model | extract |
fix-altloc |
Resolve PDB alternate conformations | fix-altloc |
add-elem-info |
Repair PDB element columns (77–78) | add-elem-info |
opt |
Geometry optimization (L-BFGS / RFO) | opt |
tsopt |
TS optimization (Dimer / RS-P-RFO) | tsopt |
path-opt |
MEP via GSM or DMF | path-opt |
path-search |
Recursive MEP search with refinement | path-search |
scan / scan2d / scan3d |
1D / 2D / 3D bond-distance scans | scan · scan2d · scan3d |
freq |
Vibrational analysis + thermochemistry | freq |
irc |
IRC (EulerPC) | irc |
dft |
Single-point DFT (GPU4PySCF / PySCF) | dft |
sp |
Single-point MLIP energy / forces / Hessian | sp |
bond-summary |
Compare structures, report bond changes | bond-summary |
trj2fig / energy-diagram |
Energy plot / R→TS→P diagram | trj2fig · energy-diagram |
pdb2reaction --help # top-level
pdb2reaction <subcmd> --help # core options
pdb2reaction <subcmd> --help-advanced # full option setIssues: https://github.com/t-0hmura/pdb2reaction/issues.
@misc{ohmura2026pdb2reaction,
author = {Ohmura, Takuto and Sato, Hajime and Terada, Tohru},
title = {pdb2reaction: End-to-End Reaction-Path Elucidation from PDB Structures Using Machine-Learning Interatomic Potentials},
year = {2026}, doi = {10.26434/chemrxiv.15003538/v1}, note = {ChemRxiv preprint}
}Agent Skills for Claude Code / Codex / Cursor etc. in skills/ — copy into your project's skill location (e.g. .claude/skills/) to let an agent drive pdb2reaction workflows and subcommands.
- MACE + UMA cannot coexist (
e3nnversion conflict). Use separate conda envs. - DFT single-point cost depends strongly on basis, functional, grid, elements, and hardware; pilot one representative structure before batching.
- Every backend's TS requires an independent frequency calculation and IRC connectivity check. The pdb2reaction ORB default is fp64; independently validate frequencies and IRC results when selecting
fp32/TF32. - CPU-only execution is supported but usually much slower than GPU.
Issues and pull requests are welcome — see CONTRIBUTING.md.
GNU General Public License v3 (GPL-3.0).